Meyer–Schuster rearrangement
The Meyer-Schuster rearrangement is the chemical reaction described as an acid-catalyzed rearrangement of secondary and tertiary propargyl alcohols to α,β-unsaturated ketones if the alkyne group is internal and α,β-unsaturated aldehydes if the alkyne group is terminal.[1] Reviews have been published by Swaminathan and Narayan,[2] Vartanyan and Banbanyan,[3] and Engel and Dudley,[4] the last of which describes ways to promote the Meyer-Schuster rearrangement over other reactions available to propargyl alcohols.
When catalyzed by base, the reaction is called the Favorskii reaction.
Contents |
Mechanism
The reaction mechanism[5] begins with the protonation of the alcohol which leaves in an E1 reaction to form the allene from the alkyne. Attack of a water molecule on the carbocation and deprotonation is followed by tautomerization to give the α,β-unsaturated carbonyl compound.
Edens et al. have investigated the reaction mechanism.[6] They found it was characterized by three major steps: (1) the rapid protonation of oxygen, (2) the slow, rate-determining step comprising the 1,3-shift of the protonated hydroxy group, and (3) the keto-enol tautomerism followed by rapid deprotonation.
In a study of the rate-limiting step of the Meyer-Schuster reaction, Andres et al. showed that the driving force of the reaction is the irreversible formation of unsaturated carbonyl compounds through carbonium ions.[7] They also found the reaction to be assisted by the solvent. This was further investigated by Tapia et al. who showed solvent caging stabilizes the transition state.[8]
Rupe rearrangement
The reaction of tertiary alcohols containing an α-acetylenic group does not produce the expected aldehydes, but rather α,β-unsaturated methyl ketones via an enyne intermediate.[9][10] This reaction competes with the Meyer-Schuster rearrangement in the case of tertiary alcohols.
Use of catalysts
While the traditional Meyer-Schuster rearrangement uses harsh conditions with a strong acid as the catalyst, this introduces competition with the Rupe reaction if the alcohol is tertiary.[2] Milder conditions have been used successfully with transition metal-based and Lewis acid catalysts (for example, Ru-[11] and Ag-based[12] catalysts). Cadierno et al. report the use of microwave-radiation with InCl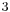 as a catalyst to give excellent yields with short reaction times and remarkable stereoselectivity.[13] An example from their paper is given below:
as a catalyst to give excellent yields with short reaction times and remarkable stereoselectivity.[13] An example from their paper is given below:
Applications
The Meyer-Schuster rearrangement has been used in a variety of applications, from the conversion of ω-alkynyl-ω-carbinol lactams into enamides using catalytic PTSA[14] to the synthesis of α,β-unsaturated thioesters from γ-sulfur substituted propargyl alcohols[15] to the rearrangement of 3-alkynyl-3-hydroxyl-1H-isoindoles in mildly acidic conditions to give the α,β-unsaturated carbonyl compounds.[16] One of the most interesting applications, however, is the synthesis of a part of paclitaxel in a diastereomerically-selective way that leads only to the E-alkene.[17]
The step shown above had a 70% yield (91% when the byproduct was converted to the Meyer-Schuster product in another step). The authors used the Meyer-Schuster rearrangement because they wanted to convert a hindered ketone to an alkene without destroying the rest of their molecule.
References
- ^ Meyer, K. H.; Schuster, K. Ber. 1922, 55, 819.(doi:10.1002/cber.19220550403)
- ^ a b Swaminathan, S.; Narayan, K. V. "The Rupe and Meyer-Schuster Rearrangements" Chem. Rev. 1971, 71, 429–438. (Review)
- ^ Vartanyan, S. A.; Banbanyan, S. O. Russ. Chem. Rev. 1967, 36, 670. (Review)
- ^ Engel, D.A.; Dudley, G.B. Organic and Biomolecular Chemistry 2009, 7, 4149-4158. (Review)
- ^ Li, J.J. In Meyer-Schuster rearrangement; Name Reactions: A Collection of Detailed Reaction Mechanisms; Springer: Berlin, 2006; pp 380-381.(doi:10.1007/978-3-642-01053-8_159)
- ^ Edens, M.; Boerner, D.; Chase, C. R.; Nass, D.; Schiavelli, M. D. J. Org. Chem. 1977, 42, 3403-3408. (doi:10.1021/jo00441a017)
- ^ Andres, J.; Cardenas, R.; Silla, E.; Tapia, O. J. Am. Chem. Soc. 1988, 110, 666-674. (doi:10.1021/ja00211a002)
- ^ Tapia, O.; Lluch, J.M.; Cardena, R.; Andres, J. J. Am. Chem. Soc. 1989, 111, 829-835. (doi:10.1021/ja00185a007)
- ^ Rupe, H.; Kambli, E. Helv. Chim. Acta 1926, 9, 672. (doi:10.1002/hlca.19260090185)
- ^ Li, J.J. In Rupe rearrangement; Name Reactions: A Collection of Detailed Reaction Mechanisms; Springer: Berlin, 2006; pp 513-514.(doi:10.1007/978-3-642-01053-8_224)
- ^ Cadierno, V.; Crochet, P.; Gimeno, J. Synlett 2008, 1105-1124. (doi:10.1055/s-2008-1072593)
- ^ Sugawara, Y.; Yamada, W.; Yoshida, S.; Ikeno, T.; Yamada, T. J. Am. Chem. Soc. 2007, 129, 12902-12903. (doi:10.1021/ja074350y)
- ^ Cadierno, V.; Francos, J.; Gimeno, J. Tetrahedron Lett. 2009, 50, 4773-4776.(doi:10.1016/j.tetlet.2009.06.040)
- ^ Chihab-Eddine, A.; Daich, A.; Jilale, A.; Decroix, B. J. Heterocycl. Chem. 2000, 37, 1543-1548.(doi:10.1002/jhet.5570370622)
- ^ Yoshimatsu, M.; Naito, M.; Kawahigashi, M.; Shimizu, H.; Kataoka, T. J. Org. Chem. 1995, 60, 4798-4802.(doi:10.1021/jo00120a024)
- ^ Omar, E.A.; Tu, C.; Wigal, C.T.; Braun, L.L. J. Heterocycl. Chem. 1992, 29, 947-951.(doi:10.1002/jhet.5570290445)
- ^ Crich, D.; Natarajan, S.; Crich, J.Z. Tetrahedron 1997, 53, 7139-7158.(doi:10.1016/S0040-4020(97)00411-0)